Search
Positive expiratory pressure devices are frequently used for airway clearance in children with cystic fibrosis and tracheobronchomalacia. This study aimed to establish if electrical impedance tomography is a feasible measure to titrate pressures in non-sedated children.

COMBAT CF is one of two long-standing international trials which have resulted in new early intervention options helping to reduce progressive lung damage in kids living with CF.
Pulmonary exacerbations pose a significant clinical burden on people with cystic fibrosis (pwCF). Whether management of exacerbations should change in the context of modulator therapy is unclear. We describe the characteristics, treatment and lung function outcomes of pulmonary exacerbations requiring intravenous antibiotic therapy (PERITs) in a contemporary Australian cohort of pwCF, in an era of rapidly broadening access to modulator therapy.
Tracheobronchomalacia (TBM) is characterised by abnormal collapsibility of the trachea and bronchi, often seen in children with cystic fibrosis (CF). This study aims to determine the impact of TBM on hospital admissions in young children with CF.
Cystic fibrosis (CF) is the most common chronic, life-shortening genetic condition affecting young Australians. There is no cure but researchers are working to prevent the onset of lung disease.

Rothwell Family Fellow; Head, Airway Epithelial Research

Two researchers from The Kids Research Institute Australia’s Wal-yan Respiratory Research Centre have secured lucrative fellowships to advance cutting-edge phage therapy research for people living with cystic fibrosis (CF).

A $350,000 Cure4 Cystic Fibrosis grant is set to propel the Wal-yan Respiratory Research Centre’s Phage WA program forward, supercharging its fight against antimicrobial resistant (AMR) lung infections in people with Cystic Fibrosis (CF) using cutting-edge phage therapy.

A promising new treatment pioneered in Western Australia for people with cystic fibrosis has commenced testing in a clinical trial in the United States and Australia.
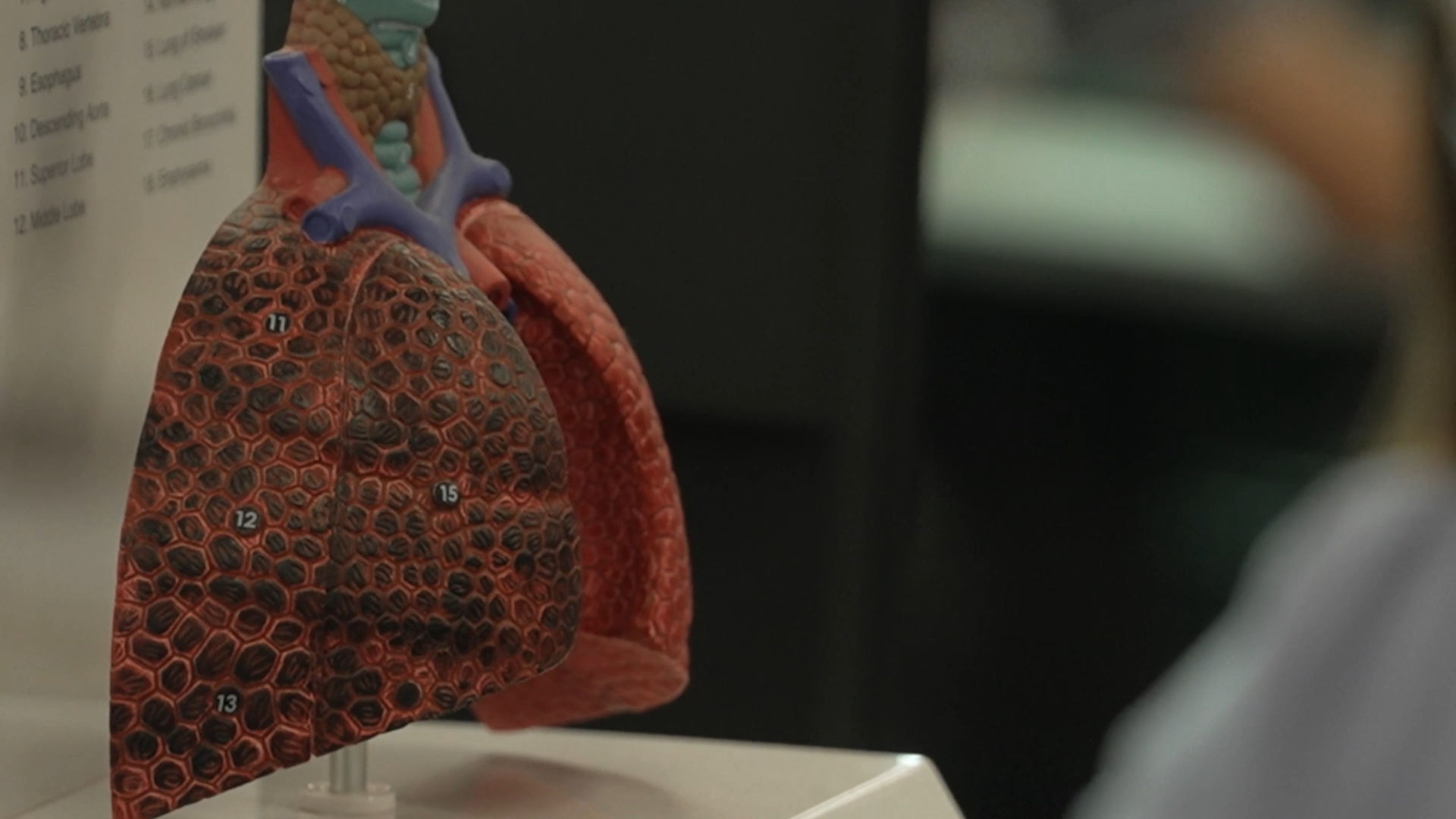
Promising results from an Australian-led clinical trial could drastically change the way we care for young children with cystic fibrosis (CF).